
Obsah [Zobrazit obsah]
Léčba spinocerebrální degenerace
Proč se spinocerebelární degenerace vyvíjí a jak se projevuje?
Spinocerebelární degenerace je skupina degenerativních onemocnění. Jejich hlavním projevem je progresivní ataxie, tedy neschopnost koordinace dobrovolných pohybů. Tato skupina onemocnění je způsobena degenerací mozečku, mozkového kmene, míchy a v některých případech periferních nervů. Mezi příčinami vědci nazývají hlavní dědičnost, ale existují případy, které nejsou způsobeny dědičnými faktory.
Co je spinocerebelární degenerace?
Spinocerebelární degenerace se vyvinou při poškození struktur centrálního nervového systému. Tyto léze jsou progresivní, což znamená, že se časem zhoršují. Vše začíná postupně. Při rychlé chůzi nebo běhu se nepostřehnutelně objevuje nemotornost nebo nestabilita. Tato neobratnost se zvyšuje a vede k nestabilitě chůze pacienta. Dále dochází k chvění končetin při provádění různých akcí a je narušena koordinace pohybů rukou. To nevyhnutelně vede ke zhoršení rukopisu, který se zvětšuje a písmena jsou nerovnoměrná.

Pokračující degenerace až atrofie nervových struktur a oblastí mozku se odráží v řeči. Stává se neostrým, méně čitelným a srozumitelným. Při změně pohledu pacienta pacient trhavě pohybuje očními bulvami, to znamená, že v určité fázi se objevují poruchy okohybnosti. Mohou se objevit i poruchy zraku s jeho oslabením až slepotou.
Proces pomalu přebírá všechny systémy těla. Je narušeno polykání a činnost trávicího traktu a močového měchýře, dochází k ochrnutí končetin, při potlačení nebo vymizení normálních reflexů se objevují patologické reflexy. Postupuje úbytek svalové hmoty doprovázený kontrakcí některých svalových vláken, což způsobuje tzv. podkožní flutter. Poměrně často spinocerebelární ataxie ovlivňuje duševní procesy a vede k degradaci osobnosti a demenci. Paralelně dochází ke snížení imunity, což přispívá k častým infekčním procesům. Přestože komplex projevů spinocerebelární ataxie vede pacienta k hluboké invaliditě, příčinou smrti jsou nejčastěji infekční komplikace.
Příznaky spinocerebelární degenerace:
- Nedostatek koordinace pohybů,
- Nejistá chůze
- Zhoršení řeči,
- zrakové postižení,
- Svalové poruchy
- Poruchy reflexu a nervového vedení
- Psychické odchylky,
- kardiomyopatie,
- Poškození imunity.
Typy spinocerebelární degenerace
I přes svou dědičnost se tento typ degenerace nejčastěji začíná projevovat nejdříve ve 20 letech. První projevy jsou obvykle pozorovány mezi 20 a 60 lety, i když některé typy těchto ataxií mohou debutovat v prvních letech života.
Je zvykem rozlišovat minimálně 7 typů spinocerebelárních degenerací. Některé z nich byly pojmenovány po vědcích, kteří popsali jejich rysy a vlastnosti. Každý z těchto typů se ve větší či menší míře liší tím či oním projevem, věkem pacientů, akumulací nebo nedostatkem určitých látek a rychlostí rozvoje chorobných procesů.
Nejčastějším typem je Friedreichova ataxie. Toto onemocnění se projevuje častěji v dětství (od 2 do 15 let). Hlavní příčinou problémů v těle je mutace v genu kódujícím protein frataxin. V důsledku toho se nevytváří v potřebném množství, což způsobuje přebytek železa a volných radikálů v těle, což vede k poškození nervového systému a různých orgánů. Poměrně často se s Friedreichovou ataxií rozvíjejí endokrinní poruchy, zejména diabetes mellitus. To přináší pacientovi další problémy.
Diagnostika a léčba
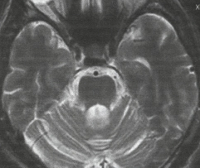
Pro stanovení správné diagnózy je nutné vyloučit onemocnění, jejichž příznaky mohou být podobné, například některé intoxikace, rakovina. Vyžaduje se počítačová tomografie a magnetická rezonance mozku. Typicky tyto studie ukazují znatelný pokles velikosti mozečku. Musí být také provedeno testování DNA.
Neexistuje účinná léčba, která by člověka zachránila před spinocerebelární ataxií nebo alespoň zastavila proces degenerace. Hlavními léky jsou Cerebrolysin a vitamíny. Nové léky ze skupiny neuropeptidů dokážou zpomalit progresi nemoci a zastavit ji na několik let. Tyto léky jsou však stále ve fázi testování.
V některých případech, zvláště když má pacient pozitivní přístup, přináší balanční terapie pozitivní výsledky. Jedná se o léčebnou metodu založenou na zpětné vazbě, kdy pacient samostatně „nutí“ svaly pracovat. Za tímto účelem existují speciální simulátory a byly vyvinuty komplexy terapeutické gymnastiky. Je lepší podstoupit takovou léčbu v nemocnici pod vedením specialisty, pak můžete výsledek konsolidovat doma. Dva nebo tři kurzy ročně, doprovázené farmakoterapií a fyzikální terapií, mohou významně zpomalit postup spinocerebelární degenerace.
Spinocerebelární ataxie

Spinocerebelární ataxie – skupina geneticky heterogenních dědičných onemocnění neurologické povahy, která se projevují různými poruchami mozečku a někdy i bazálních ganglií mozku. Příznaky tohoto stavu jsou: rozvoj ataxie a nejistá chůze, zhoršená koordinace pohybů a další neurologické projevy. Diagnóza spinocerebelární ataxie je stanovena na základě neurologického vyšetření, studia dědičné anamnézy pacienta, magnetické rezonance a molekulárně genetických studií. V současné době neexistuje žádná specifická léčba této patologie; k udržení optimální kvality života pacienta se používají metody podpůrné a symptomatické terapie.
Přehled
Spinocerebelární ataxie je skupina dědičných neurologických stavů charakterizovaných vývojem progresivní degenerace cerebelárních buněk a někdy i bazálních ganglií až do jejich úplné atrofie. Poprvé byla jedna z nemocí této skupiny popsána již v roce 1891 německým neurologem P. Menzelem, který identifikoval rozvoj ataxie, oftalmoplegie a dalších neurologických poruch ve stejné rodině. Další výzkum ukázal, že tento stav (nyní známý jako spinocerebelární ataxie typu 1) se dědí autozomálně dominantním způsobem.
V současné době se pomocí metod moderní genetiky podařilo odhalit více než 20 různých genetických variant tohoto onemocnění, přičemž více než 90 % všech případů je způsobeno pouze 6 z nich (typy 1, 2, 3, 6 , 7 a 8). Všechny formy spinocerebelární ataxie jsou charakterizovány autozomálně dominantní dědičností s jevy očekávání (zvyšování závažnosti patologie z generace na generaci) a „otcovským přenosem“ – živějším klinickým obrazem onemocnění, když je zděděno od otce. Proto je v řadě regionů v obecné struktuře patologie pozorována mírná převaha mužských pacientů. Celkový výskyt spinocerebelární ataxie se velmi liší (1-24:100 000), přičemž 1. typ je běžný v Rusku a většině Evropy, 2. typ v Indii, 3. typ v Německu a Japonsku.
Příčiny a klasifikace spinocerebelárních ataxií
Přes významnou genetickou a částečně klinickou diverzitu spinocerebelárních ataxií jsou molekulární mechanismy genetických poruch u těchto onemocnění velmi podobné. Hlavní příčinou patologie je změna počtu trinukleotidových sekvencí (CAG) v kódující části genů spojených s onemocněním. To vede ke zvýšení množství aminokyseliny glutaminu ve výsledném proteinu, což mění fyzikálně-chemické vlastnosti proteinu a zhoršuje jeho funkce. V některých případech se výše uvedené proteiny přímo či nepřímo podílejí na metabolismu nervové tkáně, takže změny v jejich struktuře vedou ke spinocerebelární ataxii. V současné době jsou nejlépe studovány molekulární mechanismy 6 hlavních typů tohoto onemocnění – tyto formy patologie jsou nejčastější a dohromady tvoří více než 90 % případů spinocerebelární ataxie.
Spinocerebelární ataxie typu 1 je považována za nejběžnější a nejvíce studovanou variantu této patologie. Je způsobena mutacemi v genu ATXN1, který se nachází na 6. chromozomu. Normálně tento gen nemá více než 36 CAG repetic; zvýšení jejich počtu vede k rozvoji onemocnění. Produktem exprese genu ATXN1 je speciální protein vázající DNA, který se aktivně podílí na metabolismu Purkyňových buněk mozečku – v přítomnosti mutantní verze genu to vede ke vzniku agregátů a postupné degeneraci , která se stává příčinou spinocerebelární ataxie.
Spinocerebelární ataxie typu 2 – méně častá varianta onemocnění, etiologie nebyla tak důkladně prozkoumána. Příčinou patologie je zvýšení počtu CAG repetic v genu ATXN2, lokalizovaných na 12. chromozomu. Ve zdravé verzi genu se počet výše uvedených sekvencí pohybuje od 15 do 36, zatímco u spinocerebelární ataxie jich může být přes 100. Funkce proteinu kódovaného genem ATXN2 jsou v současnosti neznámé.
Spinocerebelární ataxie typu 3 (jiný název je Machado-Josephova nemoc na počest dvou pacientů, u kterých byl tento stav poprvé popsán) - příčinou této varianty patologie jsou poruchy v genu ATXN3, lokalizovaném na 14. chromozomu. Normálně počet CAG repetic v tomto genu nepřesahuje s rozvojem onemocnění 47 až 53 repetic; Tento gen kóduje protein, který se pravděpodobně podílí na energetickém metabolismu neuronů v cerebellum a bazálních gangliích.
Spinocerebelární ataxie typu 6 – poměrně vzácný typ onemocnění způsobený defekty genu CACNA1A, lokalizovanými na 19. chromozomu. Pro rozvoj patologie stačí velmi mírné zvýšení počtu CAG repetic – pokud se v normální variantě genu nacházejí v 5-20, pak v přítomnosti ataxie – 21-26. Gen CACNA1A kóduje proteinovou podjednotku vápníkových kanálů umístěnou na cerebelárních neuronech. Kromě spinocerebelární ataxie způsobují poruchy v genu CACNA1A rozvoj epizodické ataxie a některých dědičných forem migrény.
Spinocerebelární ataxie typu 7 – tento typ patologie je způsoben poruchami struktury genu ATXN7, který se nachází na 3. chromozomu. U zdravého člověka není počet opakování CAG větší než 35, zatímco u onemocnění může jejich počet dosáhnout několika stovek. Funkce proteinu, který kóduje gen ATXN7, jsou v současnosti studovány.
Spinocerebelární ataxie typu 8 je způsobena genetickým defektem genu ATXN8, který se nachází na 13. chromozomu. Stejně jako v jiných případech je podstatou genetického defektu u tohoto stavu změna počtu CAG trinukleotidových sekvencí – obvykle jich je asi 15-50, zatímco v patologii může být počet repetic i přes 1200.
Téměř u jakéhokoli typu spinocerebelární ataxie tvoří abnormální forma proteinu, nadměrně bohatá na glutamin, depozita v jádrech nebo cytoplazmě neuronů cerebellum a bazálních ganglií ve formě hustých agregátů. Tento proces probíhá tím rychleji, čím více se počet CAG repetic v klíčovém genu liší od normy. To také vysvětluje mechanismus anticipace příznaků spinocerebelární ataxie – během procesu meiózy při tvorbě zárodečných buněk se může zvýšit počet výše uvedených trinukleotidových sekvencí, což vede ke zvýšení příznaků.
Vzhledem k tomu, že podobný jev se častěji vyskytuje při tvorbě samčích zárodečných buněk, stává se to příčinou tzv. „otcovského přenosu“, kdy je anticipace zaznamenána až při přenosu onemocnění z otce na potomka. Mnoho genetiků se domnívá, že hlavní příčina spinocerebelární ataxie nespočívá ve zvýšení „histidinových“ trinukleotidů, ale v deleci tzv. regulačních tripletů, které oddělují oblasti repetice CAG. Například u prvního typu onemocnění je to CAT, u druhého CAA – regulují počet CAG repetic a udržují stabilitu jejich počtu během meiózy.
Příznaky spinocerebelární ataxie
Přes výraznou genetickou rozmanitost spinocerebelární ataxie jsou projevy různých typů tohoto onemocnění obecně podobné a liší se pouze v drobných detailech – věk manifestace, charakteristika některých příznaků. Téměř všechny formy patologie nejsou v dětství registrovány – u dětí do 1 let byly zaznamenány pouze ojedinělé případy typu 2 a 7, průměrný věk jejich projevu je 18-30 let. Spinocerebelární ataxie typu 3, 6 a 7 se vyznačují ještě pozdějším vývojem – jejich projev se téměř vždy vyskytuje u osob starších 30 let. Často jsou podobné poruchy detekovány u starších lidí, což ztěžuje odlišení tohoto stavu od Parkinsonovy choroby a dalších neurodegenerativních onemocnění starších lidí.
Nejčastěji začíná vývoj spinocerebelární ataxie s výskytem jednoduché neobratnosti v pohybech, zejména při chůzi a běhu. Následně dochází k třesu rukou, poruchám chůze, paralýze extraokulárních svalů (oftalmoplegie) a ke změně rukopisu pacienta (zvětší se, linie jsou nerovnoměrné). V konečném důsledku vede onemocnění k těžké cerebelární ataxii, poruchám pyramidálních a extrapyramidových drah a parkinsonismu. Některé formy patologie jsou charakterizovány závažným poškozením zraku – rozvojem atrofie optického nervu, retinální pigmentové degenerace a dalších procesů.
Spinocerebelární ataxie typu 6, 7 a 8 se projevuje také poruchami řeči (dysartrie) a polykáním, což způsobuje potíže při krmení a vyčerpání pacientů. Právě tato okolnost a související poruchy (například atrofie mozečku, srdeční selhání) často způsobují smrt pacientů. V závislosti na formě onemocnění, množství podpůrné léčby a kvalitě péče o pacienta se může délka života u spinocerebelární ataxie pohybovat od 10 do 25 let od okamžiku, kdy se objeví první příznaky patologie.
Diagnostika a léčba spinocerebelárních ataxií
Spinocerebelární ataxie je identifikována na základě neurologického vyšetření, genetické anamnézy, magnetické rezonance mozku a molekulárně genetických studií. Při vyšetřování pacientů v různých fázích vývoje patologie se zjišťují neurologické poruchy různé závažnosti – třes končetin, ataxie, změny řeči a hlasu a v konečných fázích – dysfagie. Některé formy spinocerebelární ataxie jsou doprovázeny poměrně rychlým rozvojem zrakového postižení, které vede k úplné slepotě. Dlouhodobé sledování takových pacientů potvrzuje stabilně progresivní průběh onemocnění. Při studiu dědičné anamnézy lze určit charakteristické znaky spinocerebelární ataxie – autosomálně dominantní dědičnost, přítomnost anticipace při přenosu onemocnění od otce.
MRI mozku se spinocerebelární ataxií odhaluje ložiska demyelinizace a neurodegenerace v hemisférách, cerebelární vermis a bazálních gangliích. V terminálních stadiích onemocnění může dojít k úplné atrofii mozečku. Molekulárně genetické studie spinocerebelární ataxie se scvrkávaly na hledání patologicky zvýšeného počtu CAG repetic v genech spojených s tímto onemocněním. V současné době většina laboratoří na světě pátrá po tomto defektu v genech, které nejčastěji vedou k rozvoji patologie – ATXN1, ATXN2, ATXN3, ATXN7, ATXN8 a CACNA1A.
Neexistuje žádná specifická léčba této patologie, udržovací terapie může poněkud zpomalit vývoj spinocerebelární ataxie, ale v současné době neexistuje konsenzus o její účinnosti. Využívá se vitaminoterapie (E, A, skupina B), nootropika, metabolické stimulanty (riboxin) a metabolismus v nervové tkáni. Pokud se vyvinou mimovolní pohyby, doporučuje se použít klonazepam a haloperidol. Fyzikální terapie hraje důležitou roli při omezování progrese spinocerebelární ataxie – pravidelné provádění správně zvolené sestavy cviků pomáhá posilovat svaly a snižovat závažnost poruch rovnováhy. Za stejným účelem se doporučuje provádět terapeutická masážní sezení a elektromyostimulační postupy.
Prognóza a prevence spinocerebelárních ataxií
Dlouhodobě je prognóza jakékoli formy spinocerebelární ataxie nepříznivá – toto onemocnění je charakterizováno výrazným progresivním průběhem a postupem času vede nejprve k invaliditě a následně ke smrti pacienta. V konkrétním případě však může být prognóza méně negativní – například pokud se patologie rozvine ve stáří a udržovací léčba je zahájena včas, většina závažných příznaků se prostě nestihne objevit. Pokud se spinocerebelární ataxie objeví v mladém nebo dětském věku, délka života takových pacientů se i při intenzivní léčbě a pečlivé péči prudce zkrátí. Prevence je prováděna metodou lékařského a genetického poradenství rodičů, jejichž dědičná anamnéza je tímto stavem zatížena, a genetickou prenatální diagnostikou. V tomto případě je nutné vzít v úvahu autozomálně dominantní povahu dědičnosti spinocerebelární ataxie a takové rysy jejího přenosu, jako je anticipace.
Spinocerebelární ataxie: příčiny, příznaky, diagnostika, léčba

Každý den člověk provádí velké množství účelných pohybů, na jejichž realizaci se zapojují různé svalové skupiny (svaly antagonisty, svaly synergické, svaly agonisty, svaly fixační), vestibulární aparát, mozek, mícha a vizuální analyzátor.
Jedním z nejsložitějších motorických procesů v našem těle je chůze a udržování rovnováhy. Svaly trupu a končetin se důsledně stahují a uvolňují – objeví se krok, centrální nervový systém reguluje rychlost kroku, zahajuje a zastavuje chůzi, přizpůsobuje pohyb měnícím se podmínkám prostředí (výstup, sestup atd.), zrak analyzátor přenáší informace o okolním světě, vestibulární aparát se podílí na udržování rovnováhy v prostoru.
V okamžiku, kdy některý ze systémů přestane přesně řídit proces chůze nebo jiného motorického aktu, objeví se dezorganizované, špatně koordinované pohyby. Termín „ataxie“ se používá k označení tohoto stavu.
Ataxie může být statická, kdy člověk nedokáže udržet rovnováhu ve stoje, nebo dynamická, kdy je narušena koordinace pohybů.
V závislosti na formaci, ve které se léze nachází, se rozlišuje několik typů ataxie:
- vestibulární;
- kortikální;
- cerebelární;
- citlivý atd.
Spinocerebelární degenerace
Pojmem spinocerebelární degenerace je označována skupina onemocnění nervového systému, která se projevují poruchou koordinace pohybů v důsledku kombinovaného poškození mozečku, mozkového kmene a míchy. Do této skupiny patří idiopatické a hereditární spinocerebelární ataxie.
Všechny ataxie této skupiny mají společnou progresivní poruchu hybnosti a kombinované poškození struktur centrálního nervového systému.
Poškození mozečku se v klinické praxi projevuje nejistou chůzí (chůze opilce), vychýlením těla do strany, vynecháním znaménka (nedosáhne prstem na nos, zasune klíč do zámku apod.). ), nesprávné posouzení vzdálenosti k objektu. Člověk nemůže rychle provádět opačné, střídavé akce (otáčení rukou dlaněmi nahoru a dolů), začíná být zmatený, ztrácí rytmus. Při pohybu se objevuje třes v pažích nebo nohou (záměrný třes). Řeč se stává náhlou, pomalou, „sekanými frázemi“ s nesprávným umístěním důrazu. Poruchy řeči se označují jako cerebelární dysartrie a poruchy hybnosti jako cerebelární ataxie.
Dědičné spinocerebelární ataxie
Mezi všemi dědičnými onemocněními centrálního nervového systému je ataxie na druhém místě ve frekvenci po neuromuskulárních onemocněních. Dosud nebyla vyvinuta jednotná klasifikace této skupiny, protože hereditární ataxie jsou v klinických projevech velmi různorodé a někdy není jasné, zda se jedná o stejnou či jinou variantu.
Zastavme se u nejčastějších forem dědičných spinocerebelárních degenerací.
Friedreichova nemoc
Každých 120 lidí na světě je nositelem mutovaného genu, který je zodpovědný za rozvoj onemocnění. Onemocnění se přenáší autozomálně recesivním způsobem stejně často u chlapců i dívek.
Příčiny
Mechanismus vývoje ataxie je spojen s porušením syntézy proteinu frataxinu na mitochondriích, který reguluje transport iontů železa přes buněčné membrány. V důsledku mutace dochází k odumírání mitochondrií a buněk centrálního nervového systému, slinivky břišní, myokardu a dalších orgánů. V centrálním nervovém systému jsou poškozeny míšní cerebelární dráhy, hřbetní kořeny míchy a zadní a postranní sloupce míchy. V pozdějších stadiích onemocnění se na patologickém procesu podílejí jádra hlavových nervů, cerebelárních stopek, dentátního jádra a periferních nervů. Všechny tyto struktury jsou zodpovědné za koordinaci pohybů.
Příznaky
První klinické příznaky se objevují u dětí nad 10 let. Dítě začíná pociťovat potíže s pohybem ve tmě, objevuje se nejistá chůze, klopýtání a vrávorání. Postupně je narušena zvuková výslovnost (dysartrie) a koordinace pohybů v rukou, což se projevuje potížemi při psaní a změnami rukopisu. S progresí onemocnění se objevuje slabost a atrofie (smrt) svalů nohou a paží, což vede k úplné ztrátě samostatného pohybu a ponechá člověka upoutaného na lůžko. U většiny pacientů jsou zjištěny pánevní poruchy ve formě močové a fekální inkontinence. U některých pacientů dochází k atrofii zrakových nervů, která se projevuje úplnou nebo částečnou ztrátou zraku. Ne každý má demenci. V pozdějších fázích onemocnění člověk ztrácí schopnost sebeobsluhy.
Při vyšetření neurologem se odhalí důležitý příznak Friedreichovy ataxie – vymizení šlachových reflexů (kolenní, Achillovy atd.) až po úplnou areflexii.
Patologický proces zahrnuje srdce, muskuloskeletální systém a endokrinní systém. Člověk si stěžuje na bolest v oblasti srdce, dušnost a bušení srdce. Rozvíjí se kardiomyopatie. Symptomy z kardiovaskulárního systému se mohou objevit mnohem dříve než neurologické projevy.
Deformity skeletu jsou reprezentovány zakřivením páteře, „Friedreichova noha“ (vysoká klenba nohy je kombinována s hyperextenzí prstů v počátečních falangách a jejich ohnutím v terminálních falangách). Prsty jsou také náchylné k deformaci.
Endokrinní poruchy zahrnují diabetes mellitus, dysfunkci vaječníků, poruchy puberty, obezitu a další onemocnění.
Očekávaná délka života s Friedreichovou ataxií je zřídkakdy delší než 20 let. Hlavní příčinou úmrtí je rozvoj srdečního nebo plicního selhání a infekční komplikace.
Existuje atypická forma ataxie, která se vyznačuje pozdějším začátkem (30–50 let), příznivým průběhem a pomalou progresí. V této formě nejsou detekovány kardiomyopatie, endokrinní poruchy a zánik reflexů.
diagnostika
- Testování DNA je považováno za rozhodující metodu v diagnostice Friedreichovy ataxie.
- ENMG (elektroneuromyografie) je jednou z důležitých výzkumných metod, při které se zjišťuje poškození citlivých páteřních cest, zatímco motorické nervy jsou intaktní.
- MRI. Je diagnostikována atrofie (smrt) míšní substance.
- ECHO-CG, EKG a další studie zjišťují změny v srdečním svalu.
- Krevní test na cukr a hormonální profil diagnostikují endokrinní změny.
- Rentgenové snímky páteře a chodidel pomáhají identifikovat abnormality.
Diagnóza se provádí na základě údajů ze všech výzkumných metod.
Léčba
Nebyla vyvinuta žádná speciální terapie. Všechna terapeutická opatření jsou symptomatická.
- Léky (kyselina nikotinová, riboflavin, kyselina askorbová) zlepšují mitochondriální funkce.
- Fyzioterapie (elektrická stimulace).
- Masáže a cvičební terapie.
- Ortopedická opatření: vložky a boty, operace páteře.
Dědičná ataxie způsobená nedostatkem vitaminu E
Onemocnění je méně časté než Friedreichova ataxie a přenáší se autozomálně recesivním způsobem. Jiný název pro nemoc je Friedreichova ataxie s nedostatkem vitaminu E, neboli AVED syndrom.
Příčiny
Onemocnění je založeno na mutaci genu, který je zodpovědný za zabudování vitaminu E do struktury lipoproteinů s nízkou hustotou (podílí se na antioxidační ochraně buněk).
Příznaky
Podle klinických projevů je téměř nemožné odlišit ataxii s deficitem vitaminu E od Friedreichovy ataxie. První příznaky v podobě poruchy koordinace pohybů se objevují u dětí ve věku od 4 do 18 let. V této formě dochází i k poruchám řeči a zániku reflexů. Je třeba poznamenat, že poškození kardiovaskulárního, kosterního a endokrinního systému je několikrát méně časté. Blíže do 30 let ztrácí většina pacientů schopnost samostatného pohybu a péče o sebe a upoutají se na lůžko.
diagnostika
Všichni pacienti s ataxií by měli podstoupit krevní test ke stanovení sérového vitaminu E. Snížení nebo absence jeho koncentrace v krevním séru je spolehlivým markerem onemocnění.
Léčba
Celoživotní terapie vitaminem E v denní dávce 5-10 mgkg vede k vymizení symptomů a klinickému uzdravení člověka, zvláště při včasném zahájení léčby.
Autozomálně dominantní spinocerebelární ataxie
Do této skupiny ataxií patří řada na sobě nezávislých onemocnění, která jsou od sebe velmi obtížně odlišitelná klinickými projevy.
Rozvoj genetického inženýrství umožnil jejich klasifikaci do samostatných jednotek pomocí DNA diagnostiky. Dnes je studováno více než 13 genů, jejichž mutace vedou k rozvoji dominantních ataxií. Nemoci byly pojmenovány podle pořadového čísla genu, na kterém byly anomálie nalezeny: dominantní spinocerebelární ataxie typ 1, typ 2, typ 3. typ 13 atd.
Příčiny
Genová mutace vede k syntéze nerozpustných intracelulárních molekul, které způsobují buněčnou smrt. Každý typ dominantní ataxie ovlivňuje specifické nervové buňky.
Příznaky
Bez ohledu na typ ataxie se v klinickém obraze dostává do popředí poškození mozečku a jeho spojení s ostatními částmi centrálního nervového systému. Zjišťuje se cerebelární ataxie, cerebelární dysartrie a další příznaky.
Dominantní formy ataxie se od sebe liší nástupem příznaků a průběhem procesu.
- Spinocerebelární ataxie typu 1 se vyskytuje ve věku 30 až 40 let. Člověk vyvíjí nestabilní, neobratné pohyby při rychlé chůzi a běhu. Po několika letech se k symptomům připojí porucha písma a dysartrie. Chůze se stává ataxickou a při pohybu se objevují třes v rukou. Koordinace pohybů je narušena. Současně se zvyšuje svalový tonus paží a nohou, což vede ke snížení plného rozsahu pohybu v kloubech a nucené flexi končetin. S progresí onemocnění se rozvíjí demence (demence), pánevní poruchy (fekální a močová inkontinence), třes hlavy a poruchy polykání. Člověk úplně ztrácí schopnost pohybu. Smrt nastává po 10 a více letech od infekčních komplikací.
- Spinocerebelární ataxie typu 2 je v klinických projevech podobná ataxii typu 1. Tento typ se vyznačuje koordinovanými pohyby očních bulv (sakád), častěji a rychleji dochází k inhibici šlachových reflexů.
- Spinocerebelární ataxie typu 3 (Machado-Josephova choroba). Cerebelární ataxie je u tohoto typu kombinována s dalšími neurologickými příznaky: pomalejší chůze a řeč, pomalejší pohyby obličejových svalů, domýšlivé a prudké pohyby těla a končetin (dystonie), pokles horního víčka, slabost paží a nohou ( paréza), „vypoulené oči“, záškuby svalů v ústech a další příznaky.
- Spinocerebelární ataxie typu 4 se projevuje poruchami koordinace v kombinaci se senzorickou neuropatií, která se projevuje bolestí a poruchou citlivosti v oblasti postiženého nervu.
- Spinocerebelární ataxie typu 5 a 6. Tyto dva typy ataxie se objevují ve věku 50-60 let, postupují pomalu, nevedou k hluboké invaliditě člověka a zachovávají schopnost pacienta samostatně se pohybovat a postarat se o sebe. Hlavním příznakem je nejistá chůze, může se objevit i porucha řeči.
- Spinocerebelární ataxie typu 7. Charakteristickým znakem tohoto typu ataxie je kombinace poruch hybnosti s poškozením sítnice, které může vést až ke slepotě.
- Spinocerebelární ataxie typu 8 a následujících jsou špatně studovány, protože dnes byly hlášeny ojedinělé případy těchto onemocnění.
diagnostika
- CT nebo MRI (detekuje specifické změny v mozku, vylučuje jiná onemocnění).
- Testování DNA (hlavní diagnostická metoda).
Léčba
K dnešnímu dni neexistuje žádná specifická účinná terapie.
Jako pomocná léčba se používá cvičební terapie a vestibulární gymnastika.
Jiné formy dědičných spinocerebelárních ataxií
Do této skupiny patří atrofie dentatorubropallidoluis (DRPLA), epizodická (periodická, paroxysmální) ataxie, Marinescu-Sjögrenův syndrom.
Onemocnění jsou extrémně vzácná, léčba je pouze symptomatická.
Přednáška specialisty na téma Friedreichova ataxie:
Spinocerebelární degenerace – soubor změn v centrálním nervovém systému
Spinocerebelární degenerace implikuje kombinovaný koncept celého komplexu onemocnění, které jsou založeny na poruchách struktur míchy, mozkového kmene a mozečku.
Příznaky spinocerebelární degenerace
Hlavním jednotícím znakem této patologie je její dědičný původ a zvyšující se klinický obraz ataxie.

Poškození řady struktur centrálního nervového systému je progresivní, to znamená, že má tendenci se časem zhoršovat. Nástup onemocnění je však zřídka náhlý, primárními příznaky jsou určitá nemotornost při chůzi a nestabilita při běhu. Následuje ztráta koordinace a třes končetin. Rukopis takového pacienta se stává charakteristickým – nerovnoměrným, s velmi velkými písmeny.
Po nějaké době se hlas člověka začne měnit; je obtížné mu rozumět kvůli nejasné a nezřetelné řeči. Objevují se poruchy okulomotoriky, zrak se snižuje nebo úplně mizí.
S dalším vývojem patologického stavu je narušena funkce polykání a trávení potravy, mění se močení, vzniká ochrnutí nohou a rukou. Při vyšetření v této fázi je patrná převaha patologických reflexů nad normálními. Někdy je při vyšetření patrné podkožní chvění – jedná se o malá svalová vlákna, která podléhají křečovitým záškubám.
Trpí i intelektuální sféra – vzniká demence, pacient degraduje jako člověk. Není neobvyklé, že toto onemocnění má kardiomyopatii.
Prudký pokles imunitních sil těla vede k rozvoji mnoha infekčních procesů během spinocerebelární degenerace, která je zpravidla hlavní příčinou smrti pacienta.
Odrůdy
Paradoxně, přestože jsou nemoci tohoto druhu dědičného původu, začínají se projevovat až v dost pozdním věku – po 20 letech. Nejčastěji se takové odchylky nacházejí u lidí ve věku 20 až 60 let. I když řadu nemocí lze odhalit již na začátku života.
V současné době bylo objeveno a popsáno sedm typů spinocerebelárních degenerací. Rozdíl mezi každým z nich spočívá v závažnosti určitých projevů, rychlosti, s jakou k jejich procesu dochází, a věku pacienta, kdy onemocnění nejčastěji debutuje. Uvolňování nebo hromadění určitých látek může být také odlišné.
Nejčastěji se lékaři potýkají s typem degenerativních změn mozku nazývaných Friederichova ataxie. Onemocnění začíná ve věku od 2 do 15 let. Mechanismus vývoje patologie je založen na porušení produkce určitého proteinového těla v důsledku mutace genu odpovědného za jeho produkci. To vede k poškození nervových struktur a tkání volnými radikály a železem, které se v těle hromadí v nadbytku.
Stav pacienta se zvláště zhoršuje, když (což se často stává u takové diagnózy) se rozvine diabetes mellitus.
Jak se nemoc určuje?
Při stanovení diagnózy je nutné vyloučit možnost těch patologických stavů, které mohou podle klinických příznaků připomínat spinocerebelární degeneraci – syndrom intoxikace nebo rakovinný nádor. K tomu lékař doporučuje podstoupit MRI nebo CT vyšetření mozku, které může vykazovat známky atrofických změn v mozečku a dalších strukturách.

V případě potřeby dochází ke konečnému potvrzení tohoto typu onemocnění pomocí moderního výzkumu DNA.
Pomoc při spinocerebelární degeneraci
V současné době moderní medicína nemá schopnost tuto nemoc vyléčit. Použitím nootropních léků a vitamínů je možné poněkud zmírnit stav pacienta.
Skupina léků, jako jsou neuropeptidy, může poněkud zpomalit proces atrofických a degenerativních procesů v mozku, ale stále jsou ve fázi testování.
Byla vyvinuta sada cvičení cvičební terapie pomocí speciálních simulátorů, které s pozitivním přístupem pacienta dávají dobré výsledky. Měly by být zahájeny pod dohledem odborníka a poté lze výsledek konsolidovat doma.
