
Obsah [Zobrazit obsah]
Léčba Lennoxova syndromu gasteau
U pacientů s Lennox-Gastautovým syndromem (LGS) začíná epilepsie nejčastěji mezi prvním a sedmým rokem života a projevuje se tonickými, atonickými a myoklonickými záchvaty, vedoucími k mnohočetným pádům a atypickým absenčním záchvatům EEG odhalí bilaterální, i když ne; nutně symetrické, pomalé (10 Hz) s rostoucí nebo velkou amplitudou (Beaumanoir a Dravet, 1992; Arzimanoglou et al., 2004; Beaumanoir a Blume, 2005), někdy následované několika komplexy spike-wave. Stejné výboje krátkého trvání bez klinických projevů jsou zaznamenány ve fázi pomalého spánku.
Tonické záchvaty se často objevují v noci, ale jejich projevy mohou být tak mírné, že si jich nikdo nevšimne (Aicardi a Levy Gomes 1988; Yaqub 1993; Beaumanoir a Blume 2005).
) Atonické záchvaty se vyskytují u 26–56 % pacientů s Lennox-Gastautovým syndromem. Představují náhlou relaxaci svalů a jsou častou příčinou pádů u těchto dětí, ačkoli tonické a myoklonické záchvaty mohou také vést k pádům. Jsou vážným praktickým problémem, často způsobují zranění, kterým nelze zabránit nasazením dítěte na helmu. Mechanismus pádů při nemožnosti provedení polygrafického záznamu zůstává často nejasný a v těchto případech je lepší dát přednost vágnímu termínu astatický záchvat.
Většina atonických záchvatů se vyskytuje při ztrátě vědomí a není zcela jasné, zda jsou výhradně atonické, nebo jsou doprovázeny tonickými jevy. Ve většině případů jsou detekovány stejné změny EEG jako při tonických záchvatech. To je odlišuje od čistě atonických záchvatů spojených s výbuchy aktivity vrcholové vlny pozorovanými u myoklonicko-astatických epilepsií (Oguni et al., 1993, 1996, 2001a).
b) Skutečné myoklonické záchvaty mohou nastat v téměř 28 % případů, provázejí je i pády. Typ pozorovaného záchvatu závisí na věku nástupu Lennox-Gastautova syndromu. V případech časného nástupu převažují tonické záchvaty, zatímco myoklonické záchvaty a záchvaty absence jsou častější v případech pozdního nástupu (Chevrie a Aicardi, 1972, 1996).
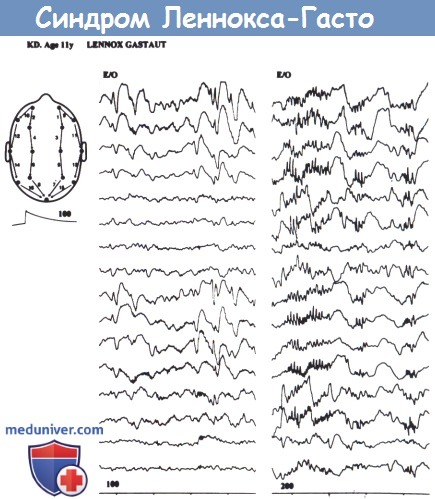
Lennox-Gastautův syndrom. EEG projevy u dítěte s různými typy záchvatů, s převahou tonických záchvatů a atypických absencí.
Fragment EEG zaznamenaný po probuzení ukazuje ostré vlny a pomalé složky asi 2-2,5/s přes přední polovinu hlavy (vlevo).
Během spánku je viditelný záblesk 10-12/sec bodových složek spojených se spontánním otevřením očí (vpravo).
c) Atypické záchvaty absence přítomna u 17–60 % pacientů. Přestože jejich nástup a konec mohou být prudší než typické záchvaty absence, vykazují stejnou ztrátu vnímání a schopnosti reagovat jemnou motorickou složkou (stupor, hypotonie, jednoduché automatismy) a jejich diagnóza závisí na klinickém kontextu a doprovodném EEG změny . Někdy během záchvatu dochází k pomalým komplexům spike-wave, obtížně odlišitelných od interiktálních spike-wave, ale častěji jsou detekovány rychlé výboje, stejně jako u tonických záchvatů.
d) Epizody se vyskytují často nekonvulzivní epistatus (Dravet a kol., 1986; Beaumanoir a kol., 1988), která může trvat několik dní nebo dokonce týdnů. Mohou způsobit střídání období zlepšení a zhoršení s výraznými změnami v reakční době a duševní aktivitě pacienta. Nejběžnější typ je charakterizován střídáním tonických záchvatů a epizod nevyzpytatelného chování, často s intermitentními myoklonickými kontrakcemi svalů obličeje a horních končetin, trvající několik hodin až týdnů (Arzimanoglou et al., 2004).
Interiktální EEG pacientů s Lennoxovým-Gastautovým syndromem vykazuje difuzní pomalý vzor hrotových vln s pomalými zkrácenými vrcholy následovanými nepravidelnými 1–2 Hz pomalými vlnami různé amplitudy, obvykle doprovázenými pomalou, nepravidelnou aktivitou pozadí. Tyto EEG projevy mohou být asymetrické a často jsou doprovázeny zjevnými klinickými projevy. Špatně nebo vůbec nereagují na hyperventilaci nebo fotostimulaci, ale jsou aktivovány během ospalosti a pomalého spánku (Aicardi a Levy Gomes, 1988).
Ve fázích nesouvisejících s REM spánkem jsou charakteristické rytmy trvající několik sekund s frekvencí 10-20 Hz, které mohou odrážet subklinické nebo minimálně výrazné tonické záchvaty. Ve stejné fázi se místo pomalých komplexů spike-wave často objevují komplexy polyspike-wave.
d) Mentální retardace před nástupem záchvatů je pozorována u 20–60 % případů (Arzimanoglou et al., 2006). Počet pacientů, u kterých se časem rozvine mentální retardace, se pět let po propuknutí onemocnění zvyšuje na 90 % (Chevrie, Aicardi 1972). Někteří pacienti mají jasnou ztrátu dovedností. Často jsou přítomny psychotické příznaky.
(e) Etiologie Lennox-Gastautova syndromu heterogenní. Důležitou roli hraje poškození mozku, zatímco genetické faktory jsou považovány za méně důležité. Až dvě třetiny případů mohou být výsledkem detekovatelných mozkových abnormalit nebo se mohou vyvinout u pacientů s opožděným vývojem v anamnéze; v druhém případě jsou považovány za symptomatické. Značný počet případů Lennox-Gastautova syndromu se vyvine po kojeneckých spasmech s postupným přechodem a jsou způsobeny stejnými mozkovými lézemi jako kojenecké spasmy.
Malformace mozku jsou však méně časté u Lennox-Gastautova syndromu a Lennox-Gastautův syndrom je extrémně vzácný u Aicardiho syndromu a lissencefalie. Často se vyskytují fokální nebo multifokální anomálie kortikálního vývoje a byly hlášeny případy heterotopie pásu a bilaterálního perisylvického syndromu. Onemocnění může být způsobeno tuberózní sklerózou a vzácnějšími neurokutánními syndromy, jako je lineární mazový névus a Ito hypomelanóza (případy z osobní praxe). Získané destruktivní léze jsou také neobvyklé. Metabolické poruchy jsou extrémně vzácné, ačkoli Lennox-Gastautův syndrom byl popsán v Leighově encefalomyelopatii (Matsuishi et al., 1985). Existují vzácné případy Lennox-Gastautova syndromu, který se vyvinul sekundárně po nádorech mozku (Honda et al., 1985).
V některých případech není zjevná příčina onemocnění, jsou považovány za kryptogenní. Kvůli četnosti Lennox-Gastautova syndromu u pacientů s jednostrannými lézemi byly učiněny pokusy rozlišit mezi „skutečnými“ Lennox-Gastautovými syndromy a případy bilaterální sekundární synchronie (Gastaut a Zifkin, 1988). Případy sekundární bilaterální synchronizace totiž splňují obvyklá kritéria pro diagnózu Lennox-Gastautova syndromu, i když jejich izolace může být užitečná z hlediska možnosti chirurgické léčby. PET zobrazování identifikovalo několik metabolických vzorců (fokální, multifokální nebo difúzní), které mohou odpovídat různým mechanismům onemocnění (Chugani a kol., 1987; Iinuma a kol., 1987; Theodore a kol., 1987). Jejich praktický význam zůstává nejasný, protože diagnostická kritéria pro Lennox-Gastautův syndrom se napříč studiemi liší.
g) Diagnóza Lennox-Gastautova syndromu nečiní žádné potíže při použití jasných kritérií. Odlišení LGS od jiných pádových syndromů však může být problematické (diskuze viz Aicardi, 1996), zejména v časných stádiích onemocnění nebo v případech, kdy ještě nejsou přítomny tonické záchvaty (Arzimanoglou et al., 2004). Vážným problémem mohou být vzácné případy „atypické dětské benigní epilepsie“ (Aicardi a Levy Gomes, 1992) nebo pseudo-Lennox syndromu (Hahn, 2000), kdy opakované pády a difúzní paroxysmální EEG aktivita během spánku mohou naznačovat Lennox- Gastautův syndrom.
Případy těchto onemocnění mohou být relativně benigní a jejich správná diagnóza je důležitá.
h) Prognóza Lennox-Gastautova syndromu nepříznivé (Arzimanoglou, 2003). Přibližně 80 % pacientů má nadále záchvaty a v závislosti na povaze záchvatů a mentálním úpadku je jen několik z nich schopno samostatného života. Výsledek je zvláště špatný u pacientů s poškozením mozku, časným nástupem nebo dřívějšími infantilními křečemi a mentální retardací před nástupem záchvatů. Normální úroveň duševního vývoje je zachována u méně než 10 % pacientů. Je obtížné odlišit tyto případy od těch s méně příznivým výsledkem, ačkoli pozdní věk nástupu, pozitivní reakce na hyperventilaci a vyšší výskyt 3Hz komplexů vrchol-vlna mají určitou prognostickou hodnotu.
Případy s takovými příznaky se označují jako „intermediate petit mal“, ale identifikace této podskupiny je kontroverzní, protože diagnostická kritéria zůstávají stejná jako u Lennox-Gastautova syndromu obecně. V typických případech se klinický obraz mění s věkem. Mezi 15. a 20. rokem se celková frekvence záchvatů obvykle snižuje. Atypické záchvaty absence a záchvaty poklesu se stávají vzácnými, ale všechny ostatní typy záchvatů přetrvávají, včetně tonických záchvatů během spánku. Je pravděpodobné, že rodiče starších dětí si méně často všimnou tonických záchvatů během spánku (Arzimanoglou et al., 2004; Beaumanoir a Blume, 2005).
i) Lennox-Gastautův syndrom je obtížně léčitelný léčba. Z moderních antiepileptik jsou kombinace valproátu sodného a benzodiazepinů neúčinné, přesto stojí za pokus tyto léky předepisovat vybraným pacientům. Někteří lékaři uvádějí, že karbamazepin je účinný u parciálních a tonických záchvatů, ale nemá žádný účinek nebo dokonce zhoršuje jiné typy záchvatů. Vzhledem k nedostatečné interakci s jinými antiepileptiky (AED) byl vigabatrin s určitým úspěchem používán, zejména u pacientů s Lennox-Gastautovým syndromem, kteří mají fokální záchvaty kombinované s nukleárními záchvaty syndromu. Bylo popsáno, že lamotrigin je účinný (Motte et al., 1997), zejména u atonických záchvatů; to může být v tuto chvíli nejlepší volba, zvláště v kombinaci s valproátem sodným.
Felbamát, užívaný v souladu se současnými doporučeními a za průběžného sledování, je často účinný i přes svou možnou toxicitu (Pellock et al., 2006). Topiramát byl hlášen jako účinný v některých případech (Sachedo et al., 1999). Je zajímavé, že je často vyžadována kombinace, protože jednotlivé léky jsou účinné pouze proti určitým typům záchvatů. Většina léčebných režimů je založena na klinických zkušenostech nebo datech z nekontrolovaných studií a pouze několik, včetně felbamátu, topiramátu a lamotriginu, je založeno na kontrolovaných studiích (Glauser a Morita 2001; Hancock a Cross 2003). Jsou vyžadovány spolehlivé informace o interakci AED; Obvykle takové děti účinněji léčí dětský neurolog, specialista na epilepsii. Nedávný Cochranův přehled (Hancock a Cross, 2003) také dospěl k závěru, že optimální léčba Lennox-Gastautova syndromu dosud nebyla nalezena a žádné studie neprokázaly, že by jakýkoli lék byl vysoce účinný. Dokud nebude proveden další výzkum, kliničtí lékaři budou muset i nadále předepisovat léčbu individuálně pro každého pacienta, přičemž zvažují možnost zlepšení oproti riziku možných vedlejších účinků.
Vzhledem k tomu, že medikamentózní terapie je často neúčinná, může být vhodné vyzkoušet jiné způsoby léčby. Ketogenní dieta má dobré krátkodobé účinky (Kinsman et al., 1992), a to i přes praktické obtíže způsobené její nechutností a vedlejšími účinky. Kinsmanovi a jeho kolegům se však podařilo prodloužit léčbu na rok nebo dva, čímž se podařilo kontrolovat záchvaty u poloviny pacientů. Zajímavé je, že tvrdí, že po dvou letech lze dietu přerušit, aniž by to způsobilo opakování záchvatů. Steroidy se používají především pro pohotovostní léčbu pacienta během období těžké aktivity nebo stavu epilepsie. Byly navrženy intravenózní imunoglobuliny, ale tato metoda nebyla dostatečně testována.
Hormon uvolňující tyreotropin (Matsumoto et al., 1987) byl používán především v Japonsku. Alternativní léčbou může být také stimulace vagusového nervu; Chirurgická resekce se používá v některých případech kortikální dysplazie. Ve vybraných případech může být provedena kalosotomie ke kontrole záchvatů s pády, které jsou nejvíce invalidizujícím typem záchvatu.
Střih: Iskander Milevski. Datum zveřejnění: 3.1.2019
Lennox-Gastautův syndrom je nejtěžší formou dětské epilepsie

Lennox-Gastautův syndrom je vzácná a závažná forma epilepsie, jejíž příznaky začínají v raném dětství. Záchvaty jsou charakterizovány různými epileptickými záchvaty.
Tento typ epilepsie je velmi obtížně léčitelný, ale medicína vyvíjí stále nové a nové techniky pro úspěšnou léčbu tohoto onemocnění.
K nástupu onemocnění obvykle dochází ve věku od dvou do osmi let, někdy i o něco později. Pro takové děti je obtížné se učit, především kvůli opožděnému obecnému vývoji – začnou sedět, plazit se a chodit samy poměrně pozdě.
Vývojové opoždění může být střední nebo závažné spolu s kognitivní poruchou.
Fyziologický a psychický vývoj každého dítěte je individuální, proto je těžké předvídat, jak se bude dítě s tímto syndromem chovat.
Zatímco většina pacientů pociťuje časté epileptické záchvaty a další různé poruchy, v některých případech poskytuje adekvátní léčba poměrně dobré výsledky a snižuje počet záchvatů.
Poprvé byla dětská myoklonická epilepsie identifikována jako samostatný syndrom v 50. letech. minulého století a o deset let později ji neurologická komunita uznala jako samostatnou nosologickou formu.
Toto onemocnění podle různých zdrojů představuje 3 až 10 % všech případů epilepsie u dětí. Prevalence patologie je jeden až dva případy na 10 tisíc lidí. Častější u chlapců než u dívek.

Patogeneze poruchy

Přestože se první příznaky objevují u dětí ve věku od dvou do osmi let, existuje samostatná ohnisková skupina pacientů, u kterých onemocnění se projevuje ve 4-6 letech.
V některých případech se nemoc transformuje z Westova syndromu, který je diagnostikován u dětí do jednoho roku. Poté se syndrom vyvine podle jednoho ze scénářů:
- kojenecké křeče Westův syndrom je nahrazen tonickými záchvaty, přeskakováním latentní formy a přecházejícím do Lennox-Gastautova syndromu;
- dětské křeče Westův syndrom mizí a dochází ke zlepšení psychomotorického vývoje.
Komplex provokujících faktorů
Dodnes nejsou známy konkrétní příčiny tohoto typu epilepsie. Mezi rizikové faktory patří:
- kyslíkové hladovění plodu v prenatálním období;
- léze mozku dítěte během prenatálního a natálního období — předčasný porod, fyziologický nedostatek;
- infekční mozkové léze kvůli zarděnkám, encefalitidě, meningitidě;
- kortikální dysplazie — porušení struktury mozkové kůry;
- tuberózní skleróza – nezhoubné nádory v mnoha tkáních a orgánech.
- idiopatický faktor – nemoc se vyvíjí z neznámých důvodů;
- genetická predispozice.
Charakteristika záchvatů
Děti s Lennox-Gastautovým syndromem mají časté a těžké epileptické záchvaty. Klinický obraz onemocnění je následující:

- Atonické záchvaty. Dochází k náhlému poklesu tonusu na několik sekund a může být pozorována i krátkodobá porucha vědomí. V případě minimální doby trvání záchvatu mají příznaky podobu pokrčení v kolenou, svěšení hlavy nebo i kývání při déletrvajícím záchvatu, dítě může zcela ztratit vědomí a upadnout;
- Tonické záchvaty. Svalový tonus se zvyšuje, ztuhnou. Záchvat může trvat několik sekund až několik minut. Často se vyskytují v okamžiku přechodu ze spánku do bdění. Pokud dítě v tuto chvíli nespí, může pád v důsledku ztráty vědomí.
- Absence záchvatů. Vědomí se na několik sekund „vypne“, zatímco dítě ztuhne a jeho pohled se zaměří na jeden bod. Také je pozorováno cukání očních víček. Při takových záchvatech nedochází k pádům; Velmi často takové útoky zůstávají několik let neodhaleny.
- Myoklonické záchvaty. Projevují se ve formě mimovolního chvění rukou a nohou, méně často celého těla. V tomto případě může pacient spadnout nebo spadnout předměty z rukou.
Mnoho dětí s Lennoxovou-Gastautovou chorobou má mentální retardaci a v důsledku toho potíže s učením a také kognitivní poruchy (poruchy chování), například nedostatek smyslu pro sebezáchovu, demonstrativnost a impulzivita.
Diagnóza onemocnění
Diagnóza onemocnění se provádí na základě následujících činností:
- Užívání anamnézy – v jakém věku se objevily první příznaky, jak probíhal porod, zda byli v rodině lidé s epilepsií, jak probíhal psychický a fyzický vývoj dítěte.
- Neurologické vyšetření – rozhovor s dítětem, využití speciálních testů a škál k identifikaci mentální retardace.
- Elektroencefalografie — analýza elektrické aktivity v mozku. Pacienti vykazují difuzní pomalé ostré vlny. Procedura se provádí po celý den ve stavu bdělosti a odpočinku, což umožňuje sledovat frekvenci útoků.
- MRI a CT — zkoumání struktury mozku vrstva po vrstvě s cílem zjistit poškození jeho struktur.
Léčba Lennox-Gastautovy choroby zahrnuje použití několika metod.
Lékařská terapie
Cílem terapie je snížit frekvenci záchvatů. Léky jsou vybírány individuálně s ohledem na minimální výskyt nežádoucích účinků.
Předepisují se následující antikonvulziva:

- klobazam;
- rufinamid;
- divalproát sodný;
- lamotrigin;
- topiramát;
- Depakine;
- karbamazepin;
Použití jednoho produktu často nedává požadované výsledky. Léky jsou předepsány komplexním způsobem a příjem je přísně kontrolován ošetřujícím lékařem.
chirurgická léčba
Pokud po lékové terapii nedojde k žádnému pozitivnímu účinku, chirurgická léčba se provádí různými metodami:

- Implantace stimulátoru vagusového nervu. Provádí se všitím do klíční kosti speciálního zařízení s elektrodou, která přenáší elektrické impulsy do bloudivého nervu. Tato metoda umožňuje snížit množství záchvaty. Jak ukazuje praxe, u více než poloviny pacientů se díky této metodě frekvence záchvatů znatelně snižuje.
- Implantace stimulátoru RNS pod pokožkou hlavy, která generuje elektrody do oblasti mozku. Elektrody nepřetržitě zaznamenávají elektrickou aktivitu mozku a při nástupu záchvatu stimulátor dodává elektrické impulsy, které potlačují epileptické ohnisko.
- Callosotomie – pitva corpus callosum, což je svazek nervů, který spojuje hemisféry mozku a přenáší epileptické vzruchy z jedné části mozku do druhé. Po operaci záchvaty úplně nezmizí, ale stanou se méně intenzivními, protože impulsy nejsou generovány z jedné hemisféry do druhé. Tato léčba se obvykle používá v případech nekontrolovaných epileptických záchvatů.
Vlastnosti jídla
Velmi často se spolu s dalšími terapeutickými metodami používá ketogenní dieta. Představuje snížení příjmu sacharidů a zvýšení příjmu tuků.
Dále se doporučují potraviny s nízkým glykemickým indexem, tedy takové, které snižují hladinu cukru v krvi. Patří sem: ovoce, zelenina, luštěniny, celozrnné výrobky, odstředěné mléko.
Během takové diety by měl lékař sledovat možnost snížení dávek užívaných léků.
Komplikace a prognóza

Onemocnění má ve většině případů nepříznivou prognózu. Asi 10 % pacientů umírá do věku deseti let, je to kvůli těžkým zraněním při atakách.
Téměř všechny nemocné děti mají v té či oné míře mentální retardaci, polovina pacientů není schopna sebeobsluhy.
Komplikace mohou také zahrnovat:
- přetrvávání záchvatů v důsledku rezistence na léčbu;
- přetrvávání duševní vady, která nemizí;
- porucha sociální a pracovní adaptace.
Zabránit vzniku tohoto onemocnění je nemožné. Mezi hlavní preventivní opatření v tomto případě lze považovat:
- Udržování vysoké kvality života – zdravý životní styl, plnohodnotný osmihodinový spánek, správná výživa, vyhýbání se vlivu stresových faktorů.
- Kvalifikovaná léčba po celý život. V žádném případě neměňte nebo nepřerušujte léčebný režim svépomocí.
Lennox-Gastautův syndrom
Lennox-Gastautův syndrom – samostatná forma dětské epilepsie, charakterizovaná přítomností polymorfních paroxysmů (myoklonické, atonické, tonické a absenční záchvaty) a opožděným neuropsychickým vývojem. Může mít kryptogenní povahu nebo působit jako syndrom jiných patologických stavů (cerebrální anomálie, genetická metabolická onemocnění, perinatální patologie). Lennox-Gastautův syndrom je diagnostikován typickým variabilním obrazem epileptických záchvatů a charakteristickým obrazcem elektroencefalogramu. Kromě toho se provádí MRI a CT mozku. Antikonvulzivní léčba syndromu je neúčinná a hledají se alternativní způsoby léčby. Prognóza je proměnlivá, ale ve většině případů nepříznivá.
Přehled
Lennox-Gastautův syndrom (LGS) je variantou dětské epilepsie, která je charakterizována kombinací atonických, myoklonických, tonických epileptických záchvatů a atypických absenčních záchvatů a pomalým EEG vzorem ostrovních vln. V roce 1950 byl LGS identifikován jako samostatný epileptický syndrom a v letech 1964-1966. Neurologická komunita ji uznala jako nezávislou nosologickou formu. Podle různých zdrojů představuje Lennox-Gastautův syndrom 3 až 10 % všech případů dětské epilepsie. Jeho prevalence se pohybuje v rozmezí 1-2,8 případů na 10 tis.. Poněkud častější je u chlapců. Typický věk nástupu onemocnění je od 2 do 5 let, méně často – 6-8 let. LGS je dnes závažné onemocnění s progresivním průběhem, na jehož účinnou léčbu stále doufá řada specialistů v oboru dětské neurologie a epileptologie.
Příčiny Lennox-Gastautova syndromu
Lennox-Gastautův syndrom je onemocnění, jehož etiologické faktory nebyly dosud přesně stanoveny. Je známo, že v mnoha případech je syndrom symptomatický a vzniká na pozadí genetické patologie, důsledků různých nepříznivých faktorů působících v perinatálním období a v 1. roce života. Ve většině případů však zůstává morfologický substrát onemocnění neidentifikován. Etiofaktory, které mohou vyvolat rozvoj LGS, zahrnují hypoxii plodu, intrauterinní infekce (rubeola, cytomegalie, herpes, toxoplazmóza), porodní poranění novorozenců (primárně intrakraniální), nedonošenost, asfyxie novorozenců, závažná infekční onemocnění v postnatálním období (meningitida, encefalitida). ), abnormality vývoje mozku (hydrocefalus, kortikální dysplazie, hypoplazie corpus callosum atd.), metabolické poruchy s poškozením centrálního nervového systému, některá genetická onemocnění (například tuberózní skleróza).
Ve 25–40 % případů se Lennox-Gastautův syndrom vyskytuje u dětí s rodinnou anamnézou epilepsie. Kromě toho existuje hypotéza o etiologické roli poruch imunity, včetně těch, které vznikají v důsledku očkování. V přibližně 30 % případů je LGS důsledkem vývoje Westova syndromu. Když se Lennox-Gastautův syndrom projevuje na pozadí úplné pohody ve zdraví dítěte a nepřítomnosti výše uvedených faktorů v jeho anamnéze, hovoří o kryptogenní (bez pravděpodobné příčiny) formě onemocnění. Kryptogenní varianta LGS se vyskytuje v 10-20 % případů a má příznivější průběh.
Příznaky Lennox-Gastautova syndromu
Symptomatický Lennox-Gastautův syndrom zpravidla debutuje na pozadí existujícího zpoždění v duševním a duševním vývoji. U kryptogenní formy odpovídá vývoj dítěte v době manifestace syndromu normě. LGS se vyznačuje velkou variabilitou útoků, jejich různou dobou trvání a četností.
Atonické záchvaty způsobené krátkodobou ztrátou svalového tonusu. Při jejich generalizované povaze dítě padá, tkzv. „pádový útok“. Lokální záchvaty mohou mít podobu náhlého ohnutí kolen, vypadávání předmětů z rukou, kývání hlavou atd. Charakteristickým rysem atonických epizod u LGS je jejich rychlost blesku a krátké trvání (do 5 sekund). Generalizované atonické paroxysmy LSH vyžadují odlišení od záchvatů myoklonicko-astatické epilepsie, mdloby a cévní mozkové příhody.
Myoklonické paroxysmy představují lokální svalové záškuby. Nejčastěji postihuje flexorové svaly proximálních paží, při rozšíření na dolní končetiny dochází k pádu. Vyznačují se symetrickým sériovým výskytem v obou končetinách a stereotypií. Potřeba odlišení od myoklonu u klíšťové encefalitidy a toxických lézí centrálního nervového systému; neepileptický myoklonus, který se vyznačuje nepravidelným asymetrickým myoklonem, který vzniká jako odpověď na různé smyslové podněty (zvuk, světlo, dotek) a není doprovázen změnami na EEG.
Tonické záchvaty DES se často vyskytují během spánku a vyznačují se krátkou dobou trvání (průměrná doba trvání 10 sekund). Doprovázeno ztrátou vědomí. Mohou mít generalizovaný charakter nebo se projevit ve formě tonického napětí jednotlivých svalových skupin (zadní krční, hřbetní, břišní svaly, ramenní pletenec atd.). Tonické paroxysmy jsou doprovázeny tachykardií, cyanózou obličeje, slzením, apnoe a hypersalivací. Minimální lokální záchvaty tonického charakteru může být někdy obtížné odlišit od zívání nebo protahování.
Atypické záchvaty absence spojené s částečnou poruchou vědomí. Projevují se jako dočasná „otupělost“, nedostatek jakékoli fyzické aktivity. Při krátkém trvání záchvaty absencí často lidé v okolí dítěte nepoznají. Při LPH mohou být záchvaty absencí provázeny svalovou hypotonií (atonické absence) a hypertonicitou zádových svalů (retropulzivní absence). Lennox-Gastautův syndrom je častěji než jiné typy epilepsie doprovázen záchvaty absencí – absencemi, které na sebe neustále navazují. Tento nekonvulzivní epistatus se obvykle vyskytuje po probuzení a může trvat několik hodin nebo dní.
Zpoždění psychomotorického vývoje (ZPR) je zaznamenán téměř ve všech případech LGS. Jeho závažnost závisí na formě syndromu (kryptogenní nebo symptomatické), povaze patologie pozadí centrálního nervového systému, závažnosti a frekvenci epileptických paroxysmů. Zpravidla se do popředí dostávají problémy s učením se novým dovednostem a osvojováním si nových informací. Často je pozorována agresivita, hyperaktivita, emoční nestabilita a charakterové rysy charakteristické pro autismus. Asi 50 % adolescentů s Lennox-Gastautovým syndromem nemá dovednosti sebeobsluhy. Dalších 25 % je sociálně a emocionálně nepřizpůsobených v důsledku těžké mentální retardace. Rysy chování a charakteru neposkytují možnost normálního přizpůsobení ve společnosti ani těm pacientům, jejichž mentální retardace je mírná. Normální sociální adaptace je pozorována pouze v 15 % případů.
Diagnóza Lennox-Gastautova syndromu
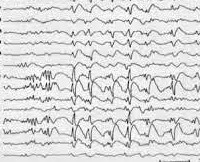
Lennox-Gastautův syndrom vzniká na základě typického klinického obrazu, skládajícího se z polymorfních epileptických záchvatů a příznaků retardovaného neuropsychického vývoje. Zohledňuje se také věk nástupu záchvatů a rodinná anamnéza epilepsie. Elektroencefalografie hraje důležitou diagnostickou roli. Interiktální (interiktální) EEG v bdělém stavu zaznamenává špatnou strukturu a pomalost základního rytmu. Vzor EEG má obraz hypsarytmie s velkým počtem hrotů různé amplitudy. Nejvyšší vrcholy jsou zaznamenány ve frontální oblasti. Vzor EEG během záchvatů závisí na jejich formě.
Neurozobrazovací metody (MRI a CT mozku) odhalují převážně nespecifické patologické změny: vnitřní hydrocefalus, atrofii subkortikálních oblastí a korových struktur především frontální zóny, hypoplazii frontálních laloků. Pokusy analyzovat pomocí PET v mozku stupeň využití glukózy mozkovými tkáněmi přinesly protichůdné informace: v některých případech byly identifikovány zóny hypermetabolismu, v jiných – hypometabolismus; U některých pacientů byl metabolismus glukózy v mezích normy.
Vzhledem k velké variabilitě paroxyzmů je třeba Lennox-Gastautův syndrom odlišit od řady dalších forem epilepsie, které se objevují v dětství: myoklonická epilepsie, benigní rolandická epilepsie, Westův syndrom, dětská absenční epilepsie, dysmetabolická epilepsie u Gaucherovy choroby, Krabbeho choroba , Niemann-Pickova choroba atd.
Léčba Lennox-Gastautova syndromu
Terapie se provádí antiepileptiky. Používá se kyselina valproová, etosuximid, karbamazepin, lamotrigin atd. Ve většině případů se kombinovaná léčba provádí jedním z těchto léčiv a valproátem sodným. Avšak až 90 % případů Lennox-Gastautova syndromu je rezistentních na antikonvulzivní léčbu. V tomto ohledu je hlavním cílem léčby snížení počtu epileptických záchvatů a zlepšení kvality života dítěte a jeho rodiny v interparoxysmálním období.
Neurologové a epileptologové hledají nové metody terapie. Je prokázána pozitivní role ketogenní diety, která spočívá v prudkém omezení konzumace sacharidů a zvýšení obsahu tuku v potravinách. Řada klinických lékařů zaznamenala pozitivní účinek léčby Lennox-Gastautova syndromu velkými dávkami imunoglobulinu. Byla pozorována účinnost ACTH a glukokortikoidů. V případech, kdy je Lennox-Gastautův syndrom provázen častými a těžkými epiparoxyzmy s pádem a hrozbou poranění dítěte, lze společně s neurochirurgem zvážit problematiku chirurgické disekce corpus callosum – kalosotomie. Takový zásah nezbavuje pacienty útoků, ale výrazně snižuje jejich intenzitu.
Mezi nové možnosti léčby patří implantace stimulátoru vagusového nervu a stimulátoru RNS. V prvním případě je zařízení instalováno subkutánně v oblasti klíční kosti a jeho elektroda je vedena do bloudivého nervu procházejícího krkem. Podle studií provedených v USA a Evropě dokáže tento přístroj v 60 % případů snížit počet záchvatů. Ve druhém případě je zařízení šito pod pokožku hlavy a jeho elektrody jsou implantovány do oblasti epileptogenního ohniska. S jejich pomocí, podobně jako EEG, přístroj neustále zaznamenává elektrickou aktivitu mozku. Při příjmu signálů indikujících nástup záchvatu generuje zařízení impulzy reakce, které zajišťují potlačení epileptické aktivity.
Prognóza Lennox-Gastautova syndromu
Lennox-Gastautův syndrom má obecně špatnou prognózu. Až 10 % případů končí smrtí dětí během první dekády života. Smrtelné následky jsou spojeny především s těžkým traumatem během epileptických záchvatů s pádem. Za prognosticky nepříznivá kritéria se považují: projevy syndromu v dřívějším věku, nástup záchvatů na pozadí mentální retardace, předchozí Westův syndrom, vysoká frekvence a intenzita záchvatů. Nemožnost lékové úlevy u epileptických záchvatů vede k progresivní mentální retardaci. Téměř všichni pacienti mají různý stupeň mentální retardace, polovina pacientů není schopna sebeobsluhy.
Vlastnosti léčby Lennox-Gastautova syndromu
Epilepsie je dnes častým problémem, jehož přesná příčina není jasná. Porucha je detekována v různém věku a má mnoho variant klinických příznaků. Jednou z nejzávažnějších forem onemocnění je Lennox-Gastautův syndrom. Je doprovázeno záchvaty a vývojovým zpožděním. Porucha je diagnostikována v dětství a většina pacientů umírá na zranění během ataky.
V současnosti dostupné léčebné metody patologie jsou v 90 % případů nedostatečně účinné. Je možné pouze snížit frekvenci a intenzitu záchvatů. S onemocněním se lze vyrovnat pouze v případech, kdy se jedná o projev jiného organického problému, pro který byly vyvinuty účinné léčebné metody.
Příčiny vzhledu
Etiologie onemocnění nebyla dosud stanovena. V medicíně se obecně uznává, že tato porucha je symptomatická. To znamená, že pouze ukazuje na přítomnost genetických abnormalit, ale tato hypotéza není podložena. Faktory, které zvyšují riziko rozvoje Lennox-Gastautova syndromu u dítěte, byly přesně identifikovány:
- Intrauterinní infekce: bakteriální a virové. Pokud je matka v těhotenství nemocná, je vysoké riziko poškození plodu. Vzhledem k tomu, že v tomto období dochází k aktivní tvorbě nervových struktur dítěte, mohou nemoci, jako je zarděnka, toxoplazmóza nebo běžný herpes, poškodit zdraví dítěte.
- Fetální hypoxie způsobená poruchami ve vývoji placenty, imunologickými problémy a dalšími poškozujícími faktory. Tento stav se může vyvinout také v prvních letech života, protože i zdravé děti pravidelně trpí apnoe – zástavou dechu, ke které obvykle dochází během spánku.
- Častou příčinou, která vyvolává rozvoj Lennox-Gastautova syndromu ve dvou až třech měsících věku, je porodní trauma. Mohou mít různé mechanismy negativního dopadu. V těžkých případech dochází k deformacím lebky a páteře, krvácením a hypoxickým jevům, které často doprovázejí zapletení pupeční šňůry kolem krku dítěte.
- Existuje genetické onemocnění, které je přímo spojeno s touto formou dětské epilepsie. Tato patologie se nazývá tuberózní skleróza a je dědičným problémem, který způsobuje poruchy nervového systému. Onemocnění je častou příčinou záchvatů.

Klasifikace a charakteristické znaky onemocnění
Lennox-Gastautův syndrom může mít odlišný klinický obraz. Záleží na typu lézí centrálního nervového systému a důvodech, které způsobily jejich vznik. Je obvyklé rozlišovat několik typů příznaků:
- Atonické záchvaty jsou doprovázeny ztrátou nebo závažným snížením svalového tonusu. Pacient není schopen ovládat vlastní pohyby. Pokud útok trvá několik sekund, může zůstat pro ostatní neviditelný, což je zvláště nebezpečné u malých dětí, které nejsou schopny mluvit o tom, co se stalo. Ztráta vědomí a pády jsou během atonie běžné.
- Myoklonické paroxysmy se vyznačují specifickými svalovými stahy, které vypadají jako záškuby končetin a trupu. Vyznačují se symetrií, to znamená, že jsou postiženy obě poloviny těla najednou. Charakteristickým rysem je stereotypní povaha pohybů pacienta. Když jsou nohy zapojeny do patologického procesu, děti i dospělí padají, což vyvolává zranění.
- Tonické záchvaty netrvají dlouho a jsou často zaznamenány v noci. To ztěžuje jejich odlišení od jiné formy epilepsie – pseudo-Lennox syndromu, který je doprovázen patologickou elektrickou aktivitou mozku během pomalého spánku (ESES – „electrical status epilepticus during slow sleep“). Na rozdíl od posledně jmenovaného není v procesu elektroencefalogramu detekován žádný specifický paroxysmus. Tonické záchvaty jsou krátkodobé. Jsou nebezpečné kvůli ztrátě vědomí, vegetativním poruchám a zástavě dechu.
- Atypické záchvaty absence jsou jedním z hlavních příznaků Lennox-Gastautova syndromu. Dochází k částečné poruše vnímání. Pacienti se na krátkou dobu stávají imobilní. Motorické projevy jsou různé: od atonie až po hypertonicitu jednotlivých svalových skupin. Když se podobné příznaky Lennox-Gastautova syndromu objeví u dětí, často si jich nevšimneme, protože trvají jen několik sekund.
- Opožděný psychomotorický vývoj doprovází mnoho duševních poruch. Představuje pacientovu neschopnost zapamatovat si nové informace a naučit se dovednosti. Děti mají potíže se zvládnutím mluveného jazyka a školní kázně. Dospělí s Lennox-Gastautovým syndromem také vykazují známky opožděného vývoje, které mají vliv i na socializaci. Takový člověk není schopen sebeobsluhy a potřebuje pomoc zvenčí. Přestože adaptace je možná, lze jí dosáhnout pouze v 15 % případů.
diagnostika
K potvrzení onemocnění je nutné komplexní vyšetření. Vzhledem k tomu, že první příznaky jsou registrovány v kojeneckém věku, je nezbytný pečlivý sběr rodinné anamnézy. Neurolog vyšetří pacienta, aby zjistil intenzitu klinických projevů. Důležitou metodou v diagnostice poruchy je elektroencefalografie (EEG), která umožňuje posoudit mozkovou aktivitu. Během záchvatů jsou výsledky specifické, i když během období normální pohody jsou zaznamenány odchylky ve fungování centrálního nervového systému. Tato metoda se také používá k rozlišení pseudo-Lennox syndromu od nočních tonických záchvatů, které mají podobné příznaky.
Protože je porucha často symptomatická, je důležité identifikovat organický základ problému, který se aktivně podílí na patogenezi. K tomu se používají vizuální metody, jako je počítačová tomografie a magnetická rezonance. Používá se také PET, který umožňuje charakterizovat metabolickou aktivitu mozku. Tato metoda však umožňuje pouze registrovat odchylky v procesu využití živného substrátu nervovými buňkami. Povaha poruch u Lennox-Gastautova syndromu je přitom odlišná. V některých oblastech mozku je zjištěn zvýšený metabolismus glukózy, v jiných je naopak snížen. V některých případech nemusí existovat vůbec žádné abnormality PET.
Konečná diagnóza je stanovena po všech nezbytných studiích a na základě shromážděné rodinné anamnézy.
terapie
Neexistují žádné účinné léčby Lennox-Gastautova syndromu. Terapie je navržena tak, aby dostala onemocnění pod kontrolu a snížila intenzitu klinických projevů. Používají se i chirurgické léčebné metody, které rovněž nejsou vysoce účinné.
Lékařská terapie
Hlavním cílem v boji proti Lennox-Gastautovu syndromu u dětí a dospělých je snížení frekvence záchvatů. K tomu se používají antiepileptika, jako je karbamazepin a fenytoin. Mají výrazný účinek pouze v 15% případů. Jejich použití je však spojeno s rizikem zhoršení stavu pacienta. Výraznější účinek má felbamát, i když patří do stejné antiepileptické skupiny léků. Umožňuje sledovat stav pacienta, ale použití léku je omezené. To je způsobeno závažnými vedlejšími účinky, které se objevují při dlouhodobém užívání. Účinné jsou léky, které jsou deriváty kyseliny valproové. Umožňují zastavit útoky, ale jejich účinek je příliš krátkodobý.
Neuroprotekce je slibná v medikamentózní kontrole mnoha typů epilepsie, včetně Lennox-Gastautova syndromu. Jedná se o poměrně mladou metodu, která je v současné době aktivně studována na modelech a zvířatech. Tato technika, i když je účinná pouze proti několika aspektům složité kaskády reakcí, které se vyskytují v mozku během neurologické poruchy, je považována za extrémně slibnou v prevenci nepříjemných následků záchvatů. Antiepileptika mají určitý ochranný účinek na nervové buňky. Neuroprotekce by však měla být zaměřena na jiné části patogeneze. Hlavní myšlenkou je ovlivňovat mechanismy spouštějící hyperaktivitu mozkových struktur, nikoli ji potlačovat.
Výzkum se zatím soustředil pouze na epilepsii posttraumatického původu, protože její povaha je nejjasnější. Léčba Lennox-Gastautova syndromu se v budoucnu může soustředit nikoli na užívání antiepileptik, ale na užívání léků snižujících negativní účinky volných radikálů. Perspektivní jsou i léky ze skupiny antagonistů NMDA receptorů.

Chirurgická intervence
Chirurgická léčba Lennox-Gastautova syndromu je založena především na kalosotomii. Tento postup zahrnuje řezání corpus callosum, struktury, která spojuje hemisféry mozku. Toto působení vede k výraznému zlepšení stavu u 40 % pacientů. Callosotomie neléčí nemoc, ale pouze zabraňuje šíření patologického vzrušení během záchvatu, což umožňuje vyhnout se pádu během paroxysmu.
Jednou z nových metod léčby syndromu je instalace stimulátoru bloudivého nervu. Takové operace se již provádějí, ale jejich účinnost se zkoumá. Callosotomy má dodnes velký úspěch. Stimulace vagu prostřednictvím instalace speciálního elektrického přístroje je zároveň spojena s nižším rizikem rozvoje problémů, zejména těch, které jsou spojené s oslabením rozumových schopností.
Prognóza a možné komplikace
Většina pacientů s Lennox-Gastautovým syndromem umírá v dětství na zranění při atakách. Je to dáno nízkou účinností v současnosti dostupných léčebných metod. Proto je prognóza onemocnění nepříznivá. I když se podaří pacienta stabilizovat, je mu přiděleno postižení. Takový člověk potřebuje neustálou pomoc a pravidelné užívání symptomatických léků. V dospělosti je syndrom spojen s výskytem dalších neurologických poruch. Pacienti mají také zvýšenou šanci na rozvoj autonomních poruch, které mohou být smrtelné.
Nutriční doporučení
Protože neexistují žádné účinné metody boje proti syndromu, hlavním cílem je zlepšit kvalitu života pacienta. Správně zvolená strava pomáhá snižovat klinické projevy. Dieta pro mnoho typů epilepsie zahrnuje snížení množství konzumovaných sacharidů a zvýšení tuků ve stravě. Zároveň mají prokázaný účinek produkty s vysokým obsahem triglyceridů se středním řetězcem. Tyto sloučeniny mají příznivý účinek na nervové struktury, díky čemuž se snižuje výskyt záchvatů. Živiny se nacházejí v kokosovém oleji, který se používá jako potravinářská přísada.
Recenze
Olga, 38 let, Soči
Prodělala těžký porod, při kterém se u dítěte rozvinula hypoxie. Lékaři miminku diagnostikovali nebezpečný typ epilepsie – Lennox-Gastautův syndrom. Začal mít záchvaty. Snažili se potíže zastavit léky, ale ve 3 měsících dítě přestalo dýchat. Nebylo možné ho zachránit.
Dmitrij, 31 let, Nižnij Novgorod
Manželka prodělala infekci během těhotenství. Když se jí narodila dcera, lékaři jí diagnostikovali Lennox-Gastautův syndrom. Byla nutná neustálá medikace. Když dítě vyrostlo, byla provedena chirurgická léčba. Četnost útoků se snížila, ale lékaři nedávají žádné uklidňující předpovědi. Doufejme v to nejlepší.